


A doença da Herança Portuguesa
A DOENÇA DA HERANÇA PORTUGUESA

A doença de descendência Portuguesa
“ A doença dos pezinhos”
Polineuropatia Amiloidótica Familiar (PAF) é uma doença genética, hereditária de evolução lenta que acomete, entre outras coisas, o sistema digestivo, com depósito de amilóide, que produz um quadro de má absorção com diarréia constante.
Geralmente estes pacientes apresentam nível de desnutrição protéica que pode ser devido à condição neuropática gastro-intestinal.
Existem pelo menos 21 tipos de amiloidose familiar.
Existem mais de 70 "mutações" de transtiretina (TTR) que estão associados a amiloidose hereditária.
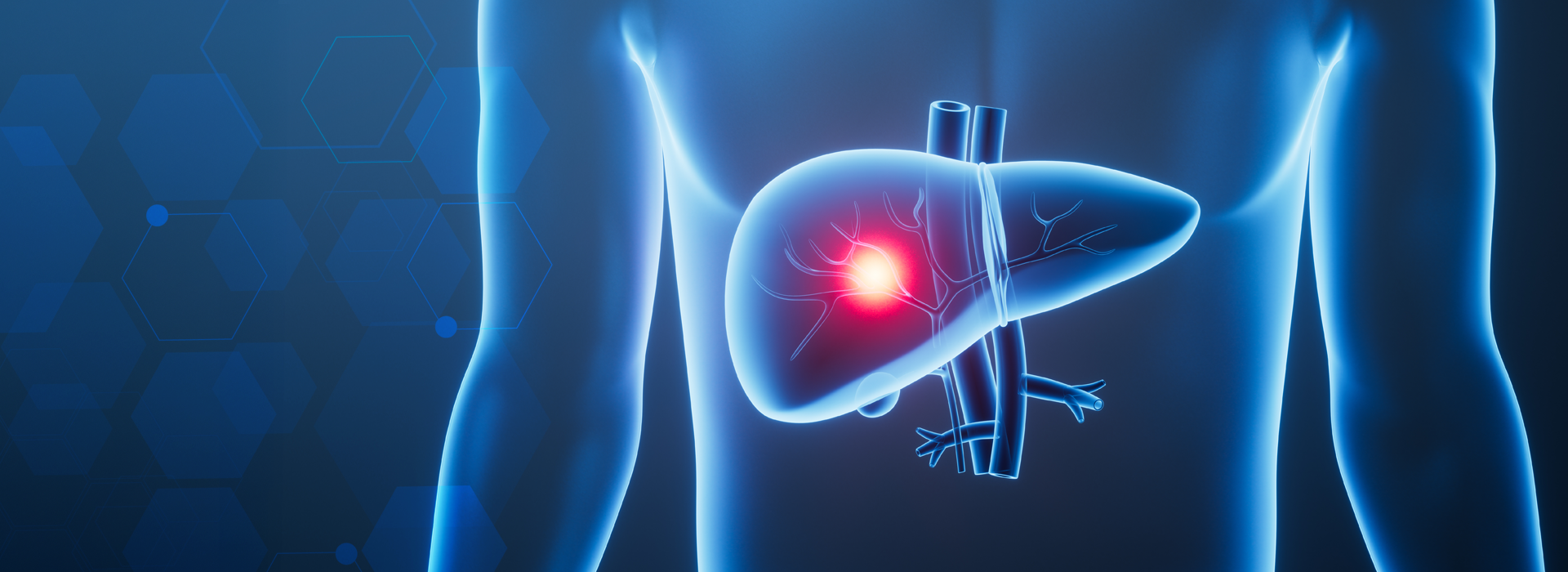
TTR é o principal constituinte dos depósitos amilóides que ocorre preferencialmente nos nervos periféricos que dão origem a polineuropatia amilóide familiar (PAF), ou no coração levando a cardiomiopatia familiar amilóide.
Isso geralmente causa dormência e formigamento nos braços e nas pernas, tonturas quando o paciente encontra-se de pé, e diarréia. No entanto, cada família tem seu próprio padrão de órgão envolvido e sintomas associados.
O modo de transmissão é autossômica dominante, o que significa que se você tiver esse tipo de amiloidose cada um de seus filhos tem 50% de chance de herdar a doença.
Se seu filho não herda o gene, ele ou ela não pode transmitir o erro metabólico às gerações futuras.
Os pacientes normalmente percebem o início dos sintomas quando adultos jovens ( após os 25 anos de idade).
Os métodos mais importantes para diagnosticar a doença são, portanto, história familiar, tecnologia do DNA e uma biópsia de nervo sural ou intestinal.
Ao examinar o DNA de uma amostra de sangue é possível determinar se um indivíduo desenvolve a doença ou não.
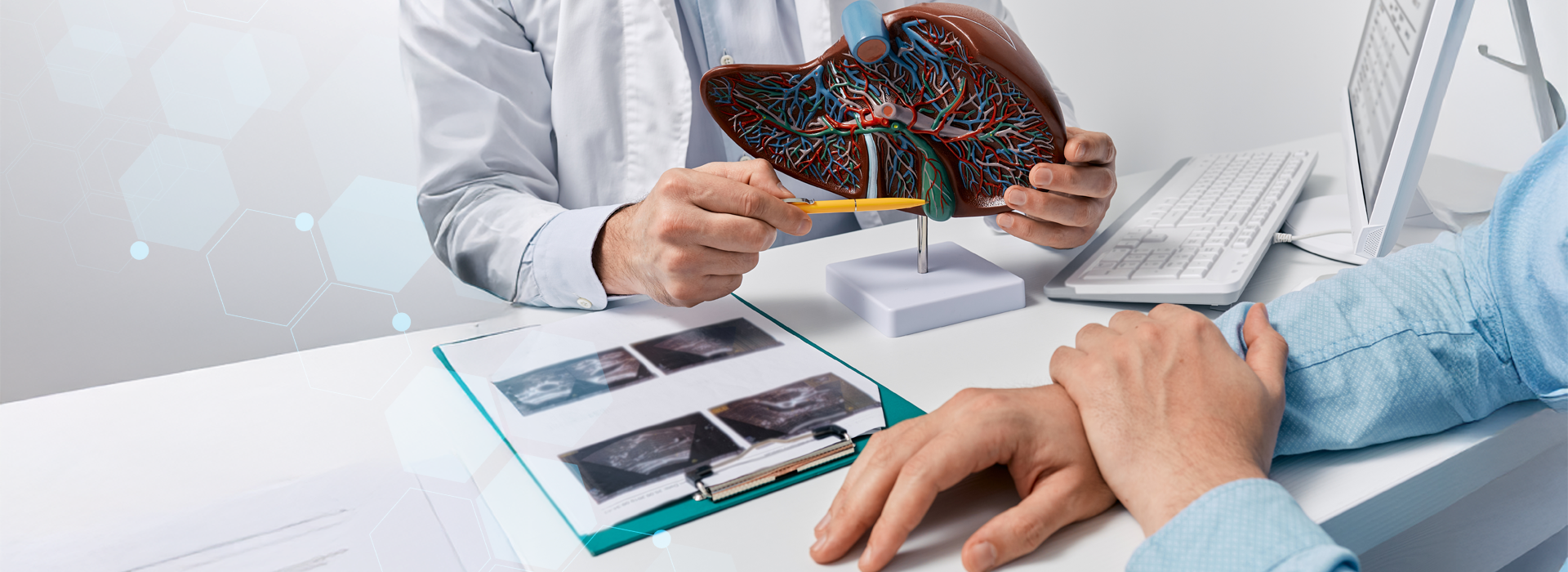
Atualmente, a única opção terapêutica eficaz é transplante hepático. Como a transtiretina familiar amiloidotica é feita no fígado, substituindo este órgão remove-se a fonte de proteína mutante produção. Um novo fígado fará apenas transtiretina normal.
Como é diagnosticada amiloidose?
Amiloidose não é uma doença comum, médicos da atenção primária muitas vezes não reconhecem quando e como deve solicitar os testes para esta doença. Nem estarão sempre atualizados sobre os últimos tratamentos.
O paciente é submetido a infinitos, morosos e dispendiosos exames, e na maioria das vezes são fechados diagnósticos de neuropatias periféricas a esclarecer.
Outros métodos diagnósticos:
1-Depósitos de amiloide no corpo:
Tecido que mostra depósitos amilóides em "coloração vermelho-Congo" é o único verdadeiro caminho para confirmar a suspeita de amiloidose.
Para obter o tecido, os meios menos invasivos devem ser utilizados em primeiro lugar, aspirado de gordura do estômago, da mucosa da cavidade oral ou retalaspiados de gordura gástrica demonstraram ser precisos em 80% nas formas hereditárias e 50% de acurácia nas formas Secundárias.
Para amiloidose hereditária, a transtiretina no sangue pode ser testada com testes de DNA.
2. Uma vez feito o diagnóstico da PAF, o que devemos fazer?
Recomendamos que você procure um Centro Médico que tenha experiência com PAF para uma avaliação.
3. A PAF pode ser curada?
Atualmente, a única opção terapêutica eficaz é transplante hepático.
4. Qual é o objetivo do tratamento?
O objetivo principal do tratamento é parar ou atrasar a produção de amilóide. Se a produção não é interrompida, o amiloide irá depositar-se no sistema nervoso periférico, e posteriormente em outras fibras neurais: autossômicas, sensitivas, cardíacas, etc.
Por este motivo o diagnóstico precoce é importantíssimo para um tratamento rápido.
Em segundo lugar, o sistema afetado, tecido ou órgão (s) devem ser tratados. O objetivo é restaurar a função, tanto quanto possível. Isto é normalmente feito com medicação, dieta, exercício e, transplante do fígado.
5. A PAF é uma doença transmissível?
Não é transmitida através do contato casual ou íntimo. É uma doença hereditária. Transmite-se através de gerações a gerações familiares.
6. O que acontece quando ocorre comprometimento da função cardíaca?
Isso significa que o paciente passa a ter "cardiomiopatia restritiva".
Nesta doença, as paredes dos ventrículos endurecem e perdem sua flexibilidade devido à infiltração de tecido anormal, tais como depósitos amilóides. Consequentemente o coração não pode encher adequadamente com o sangue e, eventualmente, perde a sua capacidade de bombear adequadamente.
A progressão da doença cardíaca gera insuficiência cardíaca congestiva: fraqueza, fadiga e sensação de sufocação. Inchaço das pernas, causada por retenção de líquidos, ocorre em um número significativo de pacientes. Outros sintomas incluem náuseas, inchaço, e perda do apetite, provavelmente devido à retenção de líquido ao redor do fígado, estômago e intestinos.
7. O que ocorre nos quadros de amiloidose renal (insuficiência renal)?
Isso significa que, em alguns pacientes, a proteína amilóide anormal depositar-se nos rins, assim, prejudicando-os.
Doença renal está presente quando os rins perdem suas funções normais causando problemas como anemia, pressão arterial elevada, por vezes necessidade de hemodiálises, doença óssea e redução da produção hormonal pelos rins.
8. Quais são as "restrições dietéticas" na Amiloidose?
Não há nenhuma restrição dietética associada à amiloidose.
Os ajustes na dieta ocorrerão de acordo com os órgãos acometidos, por exemplo: se há insuficiência renal elabora-se uma dieta que limita proteína, sódio, potássio, fósforo e líquidos. Quando um paciente tem acometimento cardíaco, a situação é diferente e o exercício pode ser mais importante. Se há comprometimento de fibras neurais intestinais, irá ocorrer diarréia constantemente e será necessário acompanhamento nutricional e dieta constipante.
9 . Medicamentos e terapias alternativas funcionam?
Muitos acreditam que uma abordagem através de medicina alternativa pode aliviar os sintomas, os doentes sentem-se melhor e terão mais força para enfrentar e aliviar o stress relacionado com aspectos da doença. Isto refere se aos sintomas que não são provenientes dos depósitos amilóides, que poderão incluir a insônia, ansiedade, as preocupações, etc
10. Tenho parentes que faleceram por essa doença ou tenho receio em imaginar que meus filhos possam herdar esta doença?
Geralmente, amiloidose é uma condição extremamente rara. As famílias sabem quando isso ocorre, pois eles têm sintomas semelhantes e as causas de morte entre os parentes de sangue normalmente acontecem em torno dos quarenta anos de idade.
Portanto, a história familiar é um indicador-chave. Se você conhecer o país de origem do seu familiar que tem ou tinha amiloidose, sobretudo se tiverem descendência Sueca, Japonêsa, Portuguesa ou italiana, deve-se investigar.
11. Testes genéticos para diagnóstico:
Testes de DNA: Dosagem de TTR-MET30 - indica o potencial para desenvolver a doença (ou seja, portadores do gene).
Então, nessas analises é possível avaliar a presença da proteína anormal, indicando se a doença está ativa. Portanto, antes do aparecimento da doença, você pode confirmar o "tipo" específico da variação da amiloidose familiar.
Se você suspeitar de amiloidose( PAF), devido sua história familiar, procure um médico e solicite ao mesmo que peça os testes de DNA para determinar se você tem a proteína amilóide variante. "Transtiretina", é o tipo mais comum de amilóide hereditária e é produzida no fígado.
16. Após o diagnóstico devemos manter qual tipo de acompanhamento?
A paramiloidose familiar torna seu portador uma pessoa com exigências e necessidades especiais do ponto de vista físico, psicológico e social.
O acompanhamento médico deverá ser por toda vida.
Saiba mais:
O primeiro órgão a falhar é, regra geral, o fígado. Não existindo tratamento eficaz, a solução passa pelo transplante hepático, procedimento que trava a progressão dos sintomas.
É uma doença congênita que acomete o sistema nervoso periférico nas suas vertentes motora, sensitiva e autonômica. Inicia-se entre os 20 e 35 anos, nos membros inferiores (por isso a designação de doença dos “pezinhos”), compromete a sensibilidade aos estímulos, a capacidade motora, sendo fatal, com evolução em média, em 10 a 15 anos.
A PAF constitui a forma mais comum de amiloidose – deposição nos tecidos, em particular nos nervos, de uma substância fibrosa (amilóide) que provoca a degeneração celular. É uma mutação característica dos doentes portugueses. Começa atrofiar os membros inferiores (daí a sua designação), assim como os superiores: pés e mãos perdem a sensibilidade térmica, sofrendo graves queimaduras. Seguem-se problemas do foro cardíaco e intestinal, enfraquecimento físico geral e, em alguns casos, impotência continuada.
Todas as manifestações da PAF traduzem a degeneração progressiva dos nervos.
No estagio final, o doente encontra-se acamado ou numa cadeira de rodas, apresentando de uma forma geral, fraqueza muscular, atrofias, e caminhando inexoravelmente para a morte por caquexia e/ou infecções intercorrentes.
A PAF é uma doença incurável e progressiva em todas as dimensões – biológica, psicológica, sociológica, cultural e espiritual - que rodeia o Ser Humano comprometido.
O calcanhar de Aquiles da genética portuguesa
Todos os dias nasce em Portugal uma criança com Polineuropatia Amiloidótica Familiar (PAF), mais conhecida por "doença do pezinho".
O primeiro órgão a falhar é, regra geral, o fígado. Não existindo tratamento eficaz, a solução passa pelo transplante hepático, procedimento que trava a progressão dos sintomas.
Tratando-se de uma doença genética, uma patologia que se manifesta sobretudo entre os 25 e 35 anos.
A PAF constitui a forma mais comum de amiloidose – deposição nos tecidos, em particular nos nervos, de uma substância fibrosa (amilóide) que provoca a degeneração celular. É uma mutação característica dos doentes portugueses. Começa atrofiar os membros inferiores (daí a sua designação), assim como os superiores: pés e mãos perdem a sensibilidade térmica, sofrendo graves queimaduras. Seguem-se problemas do foro cardíaco e intestinal, enfraquecimento físico geral e, em alguns casos, impotência continuada.
Nos últimos estágios, os órgãos mais acometidos são os rins e o coração, o que determina a morte dos pacientes.
Coube ao médico Corino de Andrade, em 1939, a descoberta da doença, ao observar que os pescadores locais não sentiam dor quando se cortavam nas cordas das redes ou se queimavam com cigarros. Foi, porém, a partir do caso de uma paciente com sintomas neurológicos distintos dos habituais que desenvolveu investigações que culminaram na primeira comunicação sobre a paramiloidose na prestigiada revista Brain, treze anos depois.
(Texto de Rosa Carreiro e Ana Tavares)
Polineuropatia Amiloidótica Familiar (PAF), Paramiloidose, Doença dos Pezinhos ou ainda Doença de Corino de Andrade
São as diversas formas de nos referirmos a esta doença descoberta pela primeira vez em Portugal, com maior frequência no norte do país. É reconhecida atualmente por todo o mundo.
Existem 4 tipos de PAF:
- PAF tipo I, de Andrade ou tipo Português
- PAF tipo II, de Rukovina ou tipo Indiana
- PAF tipo III, de Van Alien ou tipo Iowa
- PAF tipo IV, de Meretoja ou tipo Finlandês
A PAF é uma patologia neurológica, que resulta da alteração na estrutura de uma proteína – transtirretina (TTR), consequência de uma mutação no seu gene, que está localizado ao nível do cromossoma 18. È hereditária de transmissão autossômica dominante, ou seja, em caso de:
- Progenitores Homozigóticos (2 alelos mutados): os descendentes têm 100% de probabilidade de contrair a doença.
- Progenitores Heterozigótico (apenas 1 alelo mutado): os descendentes têm 50% de probabilidade de contrair a doença.
Os homozigóticos e heterozigóticos não apresentam diferenças nos sintomas, mas também pode haver homozigóticos assintomáticos. A maior parte dos doentes são heterozigóticos.
É uma doença congênita que acomete o sistema nervoso periférico nas suas vertentes motora, sensitiva e autonômica. Inicia-se entre os 20 e 35 anos, nos membros inferiores (por isso a designação de doença dos “pezinhos”), compromete a sensibilidade aos estímulos, a capacidade motora, sendo fatal, com evolução em média, em 10 a 15 anos.
Todas as manifestações da PAF traduzem a degeneração progressiva dos nervos.
Todas as fibras nervosas são atingidas e condicionam o quadro clínico da PAF:
- Fibras Sensitivas (Perda progressiva da sensibilidade, primeiro a dor e temperatura, depois tátil, vibratória e articular);
- Fibras Motoras (Atrofia muscular, Abolição dos reflexos tendinosos, Marcha em steppage);
- Fibras Autonômicas (Perturbações oculares, perturbações cardiovasculares, perturbações gastrointestinais, perturbações genito-urinárias), etc.
No estagio final, o doente encontra-se acamado ou numa cadeira de rodas, apresentando de uma forma geral, fraqueza muscular, atrofias, e caminhando inexoravelmente para a morte por caquexia e/ou infecções intercorrentes.
A PAF é uma doença incurável e progressiva em todas as dimensões – biológica, psicológica, sociológica, cultural e espiritual - que rodeia o Ser Humano comprometido.
Como medidas terapêuticas na PAF, temos:
- Marca passo – As alterações do ritmo cardíaco e da condução originam disritmias graves conduzindo inexoravelmente para a necessidade de marcapasso definitivo.
- Imunodepuração – Após o desenvolvimento laboratorial da Imunodepuração da TTR circulante no final dos anos 80, deu-se em início de 1993, a um programa de ensaios clínicos que consiste na filtração do sangue do doente, retirando a TTR met 30 através do uso de anticorpos monoclonais que se ligam à proteína anômala. ( desconheço no Brasil, o método).
- Transplante Hepático – O transplante hepático é única terapêutica definitiva, iniciada por Holmgren na Suécia em 1991 e entre nós pela Unidade de Fígado – São Paulo , com resultados encorajadores, sobretudo quando efetuado em fase relativamente precoce da doença.
O objetivo do transplante hepático reside na não evolução da sintomatologia da PAF, já que permite substituir o principal órgão produtor da proteína anômala.
No entanto, não se trata de uma terapêutica curativa.
 Twitter
Twitter
 Facebook
Facebook
 Instagram
Instagram
 LinkedIn
LinkedIn