


Hemocromatose
Hemocromatose
“A Doença Que pode nos Enferrujar”  Hemocromatose é uma doença causada pela sobrecarga (acúmulo) de ferro no organismo, que, quando atinge determinado limiar, determina lesão em vários órgãos. usualmente dividida em:
Hemocromatose é uma doença causada pela sobrecarga (acúmulo) de ferro no organismo, que, quando atinge determinado limiar, determina lesão em vários órgãos. usualmente dividida em:
Primária (também denominada Genética ou Hereditária), que é uma alteração genética que causa aumento da absorção intestinal de ferro e,
Secundária, decorrente de outras situações, como anemias crônicas, múltiplas transfusões, e outras.
O Ferro é essencial para todos os organismos vivos. Em condições normais, o conteúdo de ferro no organismo é de 3 a 4 gr.
Na Hemocromatose, a absorção pela mucosa intestinal atinge 4 mg/dia, ou mais, de ferro, com conseqüente depósito de quantidades excessivas deste metal nas células parenquimatosas, resultando em lesão tecidual e comprometimento da função de certos órgãos e glândulas.
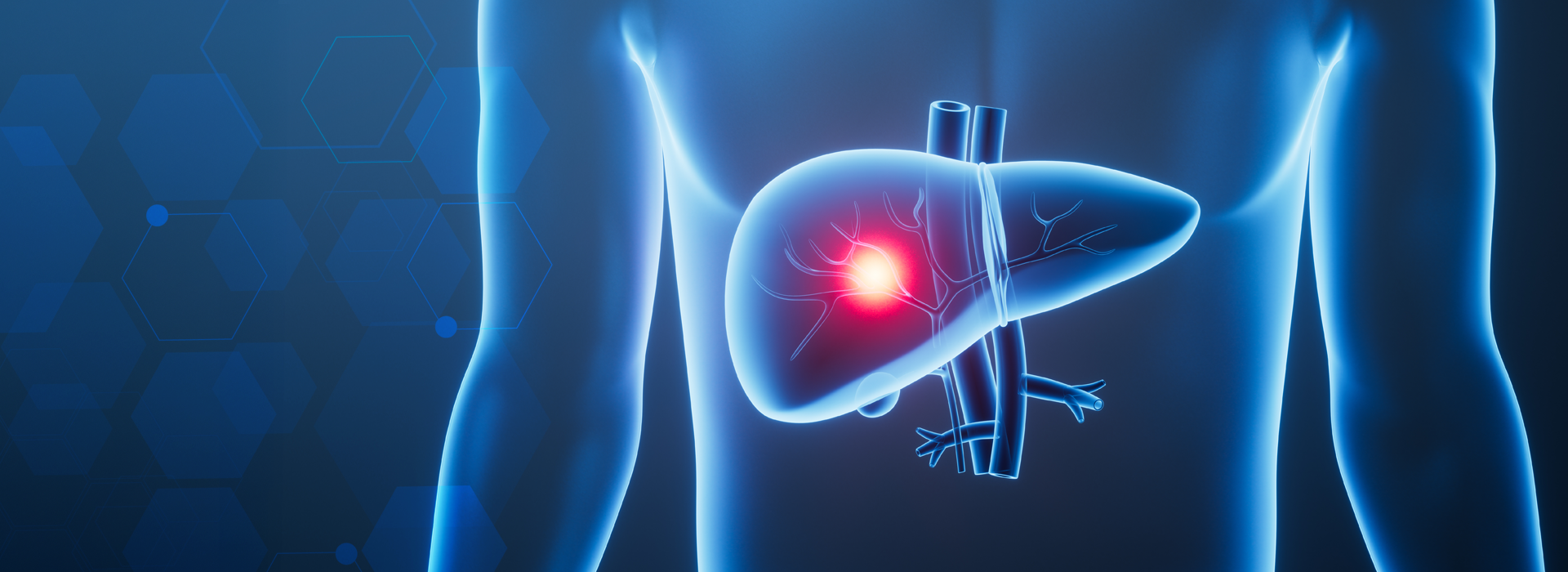
Em geral, o fígado é o primeiro órgão a ser acometido levando a aumento do órgão com evolução para esteatose (gordura no fígado), fibrose (endurecimento), cirrose e hepatocarcinoma (tumor malígno).
O pâncreas pode ser acometido pelo depósito de ferro, causando diabetes melito.
A hipófise pode ser acometida, o que causa pan-hipopituitarismo com perda dos pelos corporais, atrofia testicular, perda da libido e infertilidade.
O comprometimento cardíaco pode ser a manifestação inicial com insuficiência cardíaca congestiva, sobretudo na hemocromatose juvenil.
Há aumento difuso do coração, arritmias cardíacas e graus variáveis de bloqueio átrio-ventricular.
As artropatias com poliartrite progressiva, acometendo punhos, quadris, tornozelos e joelhos, também podem aparecer como as primeiras manifestações, dos casos em geral manifestam-se após 50 anos de idade.
Em mais de 90%, verifica-se uma pigmentação ferruginosa na pele, às vezes com tonalidade metálica ou cinza-azulada, devida ao aumento da melanina e do ferro na pele.
Nem sempre há uma correlação entre as alterações do sangue (Ferritina) e o depósito de ferro nos órgãos.
Manifestações clínicas da forma Hereditária:
Dependem do grau de sobrecarga de ferro. Nos primeiros anos de vida não há qualquer sintoma ou sinal. Após algumas décadas, quando os depósitos de ferro tornam-se elevados, começam a surgir os primeiros sintomas, que são em sua maioria muito inespecíficos e não apontam para nenhum órgão ou sistema: fadiga, fraqueza, artralgia, impotência, dor abdominal, amenorréia e perda de peso.
Com a evolução da sobrecarga de ferro, os órgãos mais acometidos são o fígado, pâncreas e coração, podendo levar a alterações graves, como insuficiência hepática, carcinoma hepatocelular, diabetes, insuficiência e arritmia cardíacas.
O acúmulo de ferro:
Em 1996 as bases moleculares da HH começaram a ser esclarecidas, através da clonagem do gene HFE (anteriormente denominado HLA-H).
A proteína HFE participa da regulação da afinidade do receptor de transferrina (proteína que transporta o ferro), que está presente em vários tipos celulares. Uma vez sintetizada, a proteína HFE forma um complexo com a 2-microglobulina e é assim transportada para superfície da célula onde se fixa em proximidade com o receptor da transferrina. Seu papel é o de regular a afinidade do receptor de transferrina, controlando assim a quantidade de ferro que é captado por cada célula.
Pesquisa das muações mais comuns:
Estudando o gene HFE em pacientes com HH, foram identificadas 2 mutações: C282Y e H63D.
A mutação C282Y torna a proteína HFE incapaz de se associar com a b 2-microglobulina, que, portanto, não é expressa na membrana celular levando a um aumento significativo e permanente da afinidade do receptor de transferrina. Na proteína com a mutação H63D, a associação com b 2-microglobulina ocorre, mas há uma perda parcial de função o que leva a aumento discreto da afinidade do receptor de transferrina.
Vários estudos em diferentes países demonstraram que cerca de 90 a 95% dos pacientes com HH apresentam homozigose para mutação C282Y, 3 a 5% apresentam homozigose para mutação H63D, são duplo heterozigotos ou heterozigotos para apenas uma das mutações.
Cerca de 1 a 3% dos pacientes não apresentam nenhuma das mutações em ambos os genes. Desta forma, ausência de mutações não exclui o diagnóstico de HH.
Diagnóstico:
Os testes bioquímicos de triagem:
1- determinação da saturação de transferrina
2- dosagem de ferritina.
Embora não haja consenso, os valores de corte para saturação de transferrina situam-se entre 45% a 55%.
Níveis de saturação acima do valor estabelecido como limite são considerados elevados, devendo-se suspeitar de HH.
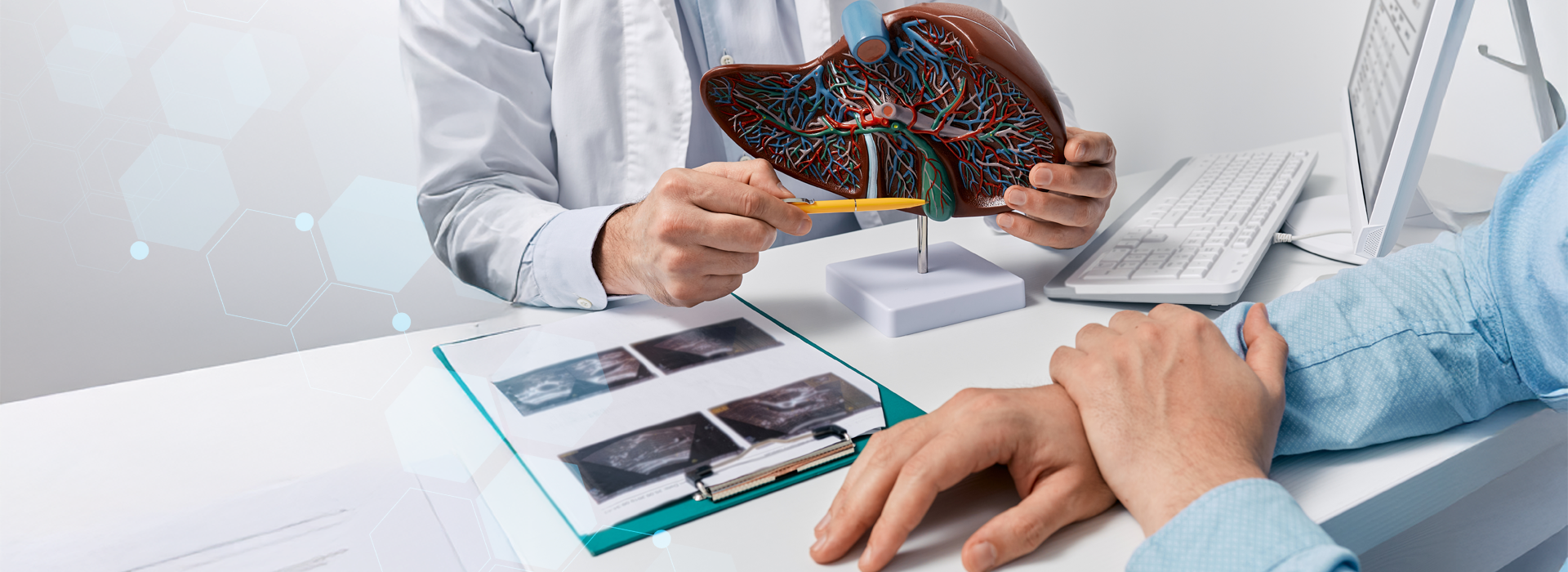
Havendo elevação concomitante dos níveis de ferritina, usualmente faz-se uma repetição destes testes e eventual prosseguimento da investigação (teste molecular, enzimas e biópsia hepáticas, exames de imagem, glicemia, avaliação cardíaca, testes hormonais, etc.).
3 - pesquisa da mutação genética para hemocromatose, Mutações C282Y e H63D p/ hemocromatose
4- biópsia hepática, mostrando hemociderose
5- Métodos de Imagem.
Complicações: Insuficiência hepática
Diabetes
Dor abdominal
Doença cardíaca
Carcinoma hepatocelular
Doença cérebro vascular
Tratamento: Consiste em eliminar o excesso de ferro e tratar as complicações. Pode ser necessário flebotomia (sangria), com a retirada de 500mls de sangue uma ou duas vezes por semana.
Uso de quelantes de ferro.
Tratamento cirúrgico: Transplante de fígado, caso cirrotização.
Hemocromatose Secundária
Ocorre pelo acúmulo de ferro, causada por outras doenças.
A sua principal causa são as hemoglobinopatias, aonde a destruição crônica de hemácias, libera a hemoglobina rica em ferro no sangue e há acúmulo nos mesmos órgãos-alvo da hemocromatose hereditária.
Outra causa estatisticamente significativa é a administração excessiva de ferro, seja pela necessidade de múltiplas transfusões sanguíneas, por auto-medicação ou pelo tratamento equivocado de uma anemia de outra causa como sendo anemia ferropriva (por deficiência de ferro).
Infelizmente, muitas vezes portadores de anemias hereditárias desenvolvem hemocromatose pelo próprio mecanismo da anemia, pela necessidade de transfusões para controlá-la e ainda fazem uso de complementos de ferro por erro diagnóstico.
Os sintomas são, na maioria dos casos : Fadiga Fraqueza Dor abdominal Perda de peso Amenorréia (ausência de menstruações) Dor nas juntas Insuficiência hepática (por cirrose e/ou carcinoma hepatocelular) Sintomas do diabetes Insuficiência e arritmia cardíacas.
Tratamento
O tratamento se baseia na retirada do excesso de ferro, por flebotomia (sangria, retirada de sangue) e/ou em uso de agentes sequestrantes de ferro, como a deferoxamina, sendo que o uso da medicação é reservada aos com contra-indicação à flebotomia, como os portadores de anemias.
Prognóstico
Com um diagnóstico precoce, pode-se evitar danos aos tecidos de diversos órgãos com a retirada do ferro em excesso.
No caso da hemocromatose hereditária, como as manifestações de sintomas são bastante difusas e relacionadas a diversas enfermidades, em muitos casos o diagnóstico ocorre por pura sorte, no momento de um check up geral, ao se observar pequenas mudanças em exames de sangue ou no rastreamento de familiares de portadores conhecidos de hemocromatose.
 Twitter
Twitter
 Facebook
Facebook
 Instagram
Instagram
 LinkedIn
LinkedIn