


Doença de Gaucher - Saiba mais
Esta doença rara e hereditária resulta de um defeito autossómico recessivo do metabolismo dos lípidos. “O serviço de remoção dos produtos celulares e gorduras não funciona. Estes resíduos acumulam-se em células exageradamente grandes, que começam a ocupar espaço e a comprometer o funcionamento dos órgãos”, explica Gabriel Tamagnini, director do Serviço de Hematologia do Hospital dos Covões, em Coimbra. Na grande maioria dos casos, a acumulação é feita no fígado e no baço, que segundo este especialista tem “tendência a “comer” as plaquetas, apresentando situações de hemorragia”.
História
Em meados do século XIX, depois de descrever o estado de uma mulher de 32 anos com o fígado e o baço muito “inchados”, o médico francês Philippe Charles Ernest Gaucher passou para a História da Medicina como o primeiro a identificar a doença de Gaucher.

Variações
A doença de Gaucher (DG) é um erro inato do metabolismo do grupo das doença lisossómicas de depósito, sendo mais frequente do referido grupo. é de herança autossómica recessiva, portanto com o risco de 25% a cada gestação de casal heterozigótico. A doença é resultante da deficiência beta-glicosidase ácida ou beta-glicocerebrosidase, que leva à acumulação de glicolípidos nos mácrofagos, principalmente no baço, fígado, medula óssea e pulmões. As manifestações clínicas ou fenotípicas da DG vão depender do grau de deficiência da enzima existindo três tipos: Tipo 1 forma não neuropática – afecta crianças e adultos com hepatoesplenomegalia, anemia, trombocitopenia, leucopenia e lesões ósseas; Tipo 2, forma neuropática aguda – afecta crianças com 4-5 meses de vida com quadro neurológico grave, hepatoesplenomegalia e comprometimento pulmonar e o Tipo 3, forma neuropática crónica – afecta crianças e adolescentes com quadro neurológico menos grave que o Tipo 2 e ainda pode comprometer o fígado, baço e ossos.
Causas
Os erros inatos do metabolismo (EIM) resultam da falta de actividade de uma ou mais enzimas específicas ou defeitos no transporte de proteínas e produzem manifestações em cada órgão desde a vida fetal à geriátrica.
A incidência a acumulativa internacional dos EIM é de 1:5000 dos recém-nascidos vivos.
A DG é um EIM do grupo das doenças lisossómicas de depósito sendo a mais frequente do referido grupo. A herança da doença é autossómica recessiva, podendo comprometer filhos de ambos os sexos. Os macrófagos com as inclusões do substrato têm uma aparência microscópica de “papel amassado” e são as chamadas células de Gaucher, que podem se encontradas na medula óssea, fígado ou baço.
A DG é resultante da deficiência de beta-glicosidase ácida ou beta-glicocerebrosidase, que leva à acumulação de glicolipídos nos macrófagos, pricipalmente no baço, fígado, medula óssea e pulmões. A DG ainda pode manifestar-se no sistema nervoso central por acumulação de metabólitos no tecido central.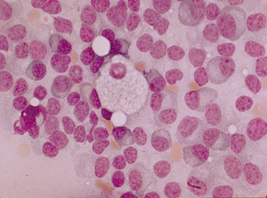
Macrófago normal entre células sanguíneas
Os macrófagos repletos de inclusões do substrato têm à microscopia a aparência de “papel amassado” e são chamadas células de Gaucher, que podem ser encontradas nos órgãos anteriormente referidos.
Hereditariedade da Doença de GaucherMais de 150 mutações no ácido desoxirribonucleico (DNA), no cromossoma 1, foram descritas, porém existem 7 alterações que correspondem à maioria dos casos. Embora não exista uma perfeita correlação entre o genótipo (tipo de mutação) e o fenótipo, pelo menos é possível distinguir a forma não neuropática e neuropática.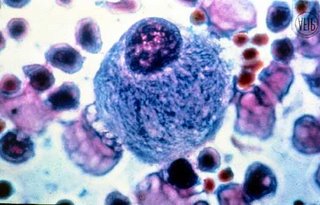 Célula de Gaucher (a maior)
Célula de Gaucher (a maior)
Sintomas
As manifestações clínicas ou fenotípicas da DG vão depender do grau de deficiência de beta-glicosidase ácida e da acumulação de glicolípidos, que são variáveis. Existem três fenótipos descritos da DG:
- Tipo 1 (forma não neuropática): Afecta crianças e adultos, a idade de início dos sinais e sintomas é muito variável. A apresentação clínica típica é hepatomegalia, esplenomegalia levando a hiperesplenismo com progressiva anemia, trombocitopenia e leucopenia. O quadro clínico é ainda associado à fadiga, cansaço, plenitude gástrica pós-prandial e atrasos de crescimento em crianças. A acumulação de glicolípidos na medula óssea leva à osteopenia, lesões líticas, fracturas patológicas, dor óssea crónica, crises ósseas, infarto e osteonecrose. É descrita também uma maior incidência de tumores ósseos nos pacientes com DG. A progressão do quadro é, em geral, lenta ou variável, e a sobrevida pode ser normal, dependendo da gravidade das complicações. O tipo 1 é o mais frequente, correspondendo a 95% dos casos de DG, tendo uma incidência de 1:10000 a 1:20000;
- Tipo 2 (forma neuropática aguda): Afecta lactentes com 4-5 meses de idade, comprometendo fígado, cérebro, baço e pulmões. O quadro neurológico é grave, com múltiplas convulsões, hipertonia, apneia e progressivo atraso mental. A incidência é menor que 1:100000. A evolução é rápida, com morte nos primeiros dois anos de vida, em geral pelo envolvimento pulmonar.
- Tipo 3 (forma neuropática crónicas): Afecta crianças e adolescentes, a idade de início é variável, mas em geral no pré-escolar. Compromete cérebro, baço, fígado e ossos. A evolução do quadro neurológico é variável, mas menos grave que o do tipo 2. A incidência é menor que 1:100000. A sobrevida dá-se até à segunda ou terceira década de vida.
Tratamento
Crianças com Doença de GaucherO tratamento desta doença é feito com base na reposição enzimática (imiglucerase) das enzimas em falta no organismo dos portadores de DG. Nos pacientes do tipo 3, o tratamento deve ser introduzido precocemente, antes do quadro neurológico grave estar estabelecido.
O cuidado deve ser muito mais intenso quando o paciente é uma criança que já apresente comprometimento ósseo, mostrando a gravidade do seu quadro; este tipo de doente merece especial atenção para a prevenção de complicações mais graves na vida adulta, que podem levar até a uma limitação incapacitante. O tratamento dos pacientes com DG bem como de outros os erros inatos do metabolismo deve ser acompanhado de uma actualização frequente sobre estas doenças pois existe um grande número de pesquisas envolvendo a qualidade e eficácia da terapêutica.
O tratamento desta doença deve incluir para além do aspecto clínico, também o aconselhamento genético periódico, suporte psicológico, fisioterápico e nutricional devido aos problemas sociais e emocionais dos portadores de DG.
 Twitter
Twitter
 Facebook
Facebook
 Instagram
Instagram
 LinkedIn
LinkedIn